A FAIR and modular image-based workflow for knowledge discovery in the emerging field of imageomics
Meghan A. Balk, John Bradley, M. Maruf, Bahadir Altintaş, Yasin Bakiş, Henry L. Bart Jr, David Breen, Christopher R. Florian, Jane Greenberg, Anuj Karpatne, Kevin Karnani, Paula Mabee, Joel Pepper, Dom Jebbia, Thibault Tabarin, Xiaojun Wang, Hilmar Lapp
First published: 22 April 2024
Posted in: A FAIR and modular image‐based workflow for knowledge discovery in the emerging field of imageomics - Balk - Methods in Ecology and Evolution - Wiley Online Library
Lead author: Meghan A. Balk.
Handling Editor: Hooman Latifi
Abstract
- Image-based machine learning tools are an ascendant ‘big data’ research avenue. Citizen science platforms, like iNaturalist, and museum-led initiatives provide researchers with an abundance of data and knowledge to extract. These include extraction of metadata, species identification, and phenomic data. Ecological and evolutionary biologists are increasingly using complex, multi-step processes on data. These processes often include machine learning techniques, often built by others, that are difficult to reuse by other members in a collaboration.
- We present a conceptual workflow model for machine learning applications using image data to extract biological knowledge in the emerging field of imageomics. We derive an implementation of this conceptual workflow for a specific imageomics application that adheres to FAIR principles as a formal workflow definition that allows fully automated and reproducible execution, and consists of reusable workflow components.
- We outline technologies and best practices for creating an automated, reusable and modular workflow, and we show how they promote the reuse of machine learning models and their adaptation for new research questions. This conceptual workflow can be adapted: it can be semi-automated, contain different components than those presented here, or have parallel components for comparative studies.
- We encourage researchers—both computer scientists and biologists—to build upon this conceptual workflow that combines machine learning tools on image data to answer novel scientific questions in their respective fields.
1 INTRODUCTION
Biological research increasingly leverages machine learning (ML) algorithms and methods (Greener et al., 2022; Mahmud et al., 2018, 2021; Tarca et al., 2007), with a focus on the growing number of biological image data resources [i.e. MorphoSource (Boyer et al., 2016), iDigBio (idigbio.org) and iNaturalist (inaturalist.org)]. Image data are ripe for applications of ML techniques, including neural networks (NN), to extract information such as metadata (Karnani et al., 2022; Leipzig et al., 2021; Pepper et al., 2021; Rinaldo et al., 2022; Stork et al., 2019), species classification (Schuettpelz et al., 2017; Wäldchen & Mäder, 2018; Wilf et al., 2016) and presence of traits (Alfaro et al., 2019; Lürig et al., 2021; MacLeod, 2017; Weeks et al., 2016). Although ML offers powerful tools for automatic object detection and subsequent analysis of biological image data, no single ML technique provides a complete solution. As a result, the need to combine various techniques to solve complex problems is inevitable.
Similar to the need to create complex, computational workflows for genomic studies generating large datasets, complex workflows are also required for computationally intensive research that uses ML to extract information from image data. We draw on lessons from the genomic world (Ahmed et al., 2021; Köster & Rahmann, 2012; Mölder et al., 2021; Papageorgiou et al., 2018) and from best practices for creating workflows (Goble et al., 2020; Leipzig et al., 2021; Shade & Teal, 2015) and apply them to the emerging field of ‘imageomics’. Imageomics harnesses revolutions in artificial intelligence and ML—as well as the rapidly growing collections of biological image data—to accelerate biological knowledge of organisms from images (About the Imageomics Institute (NSF OAC-2118240) | Imageomics Institute).
Creating FAIR (findable, accessible, interoperable, reusable), reproducible, modular, and automated workflows empowers domain scientists, the users of technologies to answer a research question, to use ML tools for their research. The need for automated and reproducible workflows that string together technologies is not unique, and has been previously discussed in biology (e.g. Brack et al., 2022; Goble et al., 2020; Haston et al., 2012; Roach et al., 2022; Shade & Teal, 2015). Although workflow tools geared for biologists who need to combine ML models on image data have been developed (Lürig, 2022; Porto & Voje, 2020; Weeks et al., 2022), biologist-oriented best practice guidelines for materializing an automated, FAIR, and reproducible imageomics workflow are, to the best of our knowledge, missing. Combining techniques and tools as FAIR components of a reusable workflow help to avoid duplication, reduce user-error, facilitate the retention of metadata and attribution, and promote reproducibility through automation. Developing workflows depends on effective collaboration among a team that includes ML researchers, who often develop the ML algorithms used as components in a workflow, and software engineers, who help create the tooling and workflows.
Here, we showcase a conceptual imageomics workflow (Figure 1). This conceptual workflow arose from a need from our interdisciplinary team to develop NNs for discovering phenotypic traits using structured biological knowledge. We recognized a need to converge on a central standardized, conceptual workflow that brings in data from shared resources, uses interoperable and portable components, and infrastructure to enable collaboration.
FIGURE 1
Open in figure viewerPowerPoint
Conceptual imageomics workflow. The general steps (row 1; grey ovals) are: (1) acquiring or downloading image metadata, (2) filtering metadata (i.e. data wrangling), (3) downloading image data, (4) applying ML models, (5) analysing outputs from the models. Components (row 2), which are scripts (pink) and tools (teal) are required to perform these steps. Each step produces an output (row 3; rectangles) that is itself read by further downstream steps or saved as a result. Together, these steps, components and tools can be called using a WM.
We implement the conceptual imageomics workflow in a specific case study (Figure 2). The application of the conceptual imageomics workflow showcases how technologies and tools can be modularized, combined and automated as an application-specific imageomics workflow definition (i.e. a workflow that has defined rules and execution).We wanted these components interoperable with different computing environments and reusable by other workflows (Brito et al., 2020; Roach et al., 2022), to be end-to-end automated for full reproducibility, and provide flexibility in how a domain scientist might configure and interact with workflow components. We follow best practices for reproducible workflows from the field of computational biology (Brito et al., 2020; Roach et al., 2022; Sandve et al., 2013), for creating FAIR and Open Science components for data, metadata, software, and ML models (Barker et al., 2022; Brito et al., 2020; Chue Hong et al., 2022; Goble et al., 2020; Jiménez et al., 2017; Miura & Nørrelykke, 2021; Roach et al., 2022; Sandve et al., 2013; Wilkinson et al., 2016), for image data reproducibility (Miura & Nørrelykke, 2021), and for the modularization of tools (Brack et al., 2022; Nüst et al., 2020). While we built our case study to be reused internally by our collaborative team, teams implementing the conceptual imageomics workflow may have different requirements for data openness and FAIR-ness. Our intention is for the conceptual workflow and the example of how to implement such a workflow using FAIR data principles to guide biological research communities using ML with image data.
FIGURE 2
Open in figure viewerPowerPoint
Application-specific imageomics workflow definition. The application-specific imageomics workflow for the case study includes steps from Figure 1 (row 1). These translate to rules for a WM (row 4; yellow) that call different components directly from external repositories (row 2; light teal), components (row 3) that are derived from containers (dark teal), and scripts (pink) from a researcher’s repository. The outputs (row 5) may feed into the subsequent rules until the final output files.
2 MATERIALS AND METHODS
2.1 Workflow
2.1.1 Conceptual imageomics workflow
The steps of a conceptual imageomics workflow (Figure 1) emphasizes findable and accessible components throughout. Each step (Figures 1 and 2, row 1) calls a component. These steps are: (1) extracting a metadata list of image data items that can be from an archive (preferable) or a local folder, (2) filtering the image data using a script, (3) downloading selected image data, (4) implementing components such as ML models, and (5) analysing the outputs using a script. The workflow does not need to be linear: outputs may be results themselves and not always feed into the next step, and/or a workflow might branch such that versions of the tools or methods can be compared.
2.1.2 Case study: Application-specific imageomics workflow definition
The purpose of this case study is to apply the conceptual imageomics workflow to a biological problem, here, the extraction of traits from images (Figure 2). We built upon our previous work (Bakış et al., 2021; Jebbia et al., 2022; Karnani et al., 2022; Leipzig et al., 2021; Pepper et al., 2021), using image data from a specific group of fishes, the minnows (Family: Cyprinidae), incorporating previously created metadata extraction methods (Karnani et al., 2022; Leipzig et al., 2021; Pepper et al., 2021), the outputs of which would become the input to a segmentation model to extract traits from image data. We chose simple traits to extract: trunk, head, eye, dorsal fin, caudal fin, anal fin, pelvic fin, and pectoral fin (e.g. Figures S1 and S2; README of Morphology-analysis repository). The ML researchers who created the components kept the associated code and models in publicly available GitHub repositories. However, as these personnel moved on from the project, the biologists (i.e. domain scientists) were unable to use or adapt the tools themselves, rendering the models inaccessible and not reusable. We recognized that integrating these computational tools into a workflow would enable the domain scientists, including new team members, to work independently and creatively.
2.2 Repositories
We use repositories to ensure that the data and components are accessible and findable to our team (Figure 2, row 2). We used the following repositories: Fish-AIR for image metadata and data, Zenodo for archiving the fish image metadata, Hugging Face for the MLmodels, and GitHub for the codebases, which include workflow definitions (see Section 7), and a registry for the containers providing the runnable workflow components (Table 1). GitHub is widely used for hosting Git-based version control repositories, including for collaborative research projects. Further, it is an application that many in the team, including the domain scientists, were familiar with and comfortable using. Likewise, we adopted Hugging Face as the ML model repository because it is widely used in the ML community, supports version control, has seamless integration with Git-based code versioning, and has rich metadata in the form of ‘model cards’. Like GitHub does for code sharing, these features enable FAIR ML model sharing, such as making it easy for researchers to find, pull and reuse the models. For permanently archiving version-specific snapshots, we used Zenodo for GitHub repositories and Hugging Face’s built-in capability to obtain DataCite DOIs for the ML models. For future reproducibility, we deposited relevant parts of the Fish-AIR metadata download in a Zenodo archive (Balk, Tabarin, et al., 2023), which is common practice among the biological (domain science) community. All resources, including repository URLs and permanent identifiers, are listed in Table 1. While there are many options for where and how to archive data, we chose these based on what our team was accustomed to using (i.e. GitHub), standards for the scientific community (i.e. Zenodo), and ease of use for the software engineers and domain scientists to use (i.e. Hugging Face).
TABLE 1. Resources used in this paper (table format adapted from Cell Press Key Resources Table).
| Resource | Source | Identifier | Citation |
|---|---|---|---|
| Deposited data | |||
| Burress et al. 2017 supplementary information | Burress et al. (2017) | https://doi.org/10.1111/jeb.13024 | Burress et al. (2017) |
| Metadata used and data generated | This paper | https://doi.org/10.5281/zenodo.10629836 | Balk, Tabarin, et al. (2023) |
| Software and algorithms | |||
| Application-specific imageomics Workflow: Minnow_Segmented_Traits | This paper | https://zenodo.org/records/8205729 |
Repository: https://github.com/hdr-bgnn/Minnow_Segmented_Traits/tree/v1.0.2|Balk, Bradley, et al. (2023)|
|Sub-workflow: BGNN Core Workflow|This paper|hdr-bgnn/BGNN_Core_Workflow: Use latest containers
Repository: https://github.com/hdr-bgnn/BGNN_Core_Workflow/tree/1.0.1|Tabarin, Bradley, Balk, and Lapp (2023a)|
|Metadata generation: drexel_metadata|Leipzig et al. (2021), Pepper et al. (2021), Karnani et al. (2022)|imageomics/Drexel-metadata-generator · Hugging Face
Container: Package drexel_metadata · GitHub
Repository: https://github.com/hdr-bgnn/drexel_metadata/tree/0.6|Karnani et al. (2023)|
|Reformat metadata: drexel_metadata_reformatter|This paper|https://doi.org/10.5281/zenodo.7987576
Container: Package drexel_metadata_formatter · GitHub
Repository: https://github.com/hdr-bgnn/drexel_metadata_formatter/tree/v0.0.1|Tabarin, Bradley, and Lapp (2023a)|
|Crop image: Crop_Image|This paper|https://doi.org/10.5281/zenodo.7987485
Container: Package crop_image · GitHub
Repository: https://github.com/hdr-bgnn/Crop_image/tree/v0.0.4|Tabarin, Bradley, and Lapp (2023b)|
|Segmentation: BGNN-trait-segmentation|This paper|imageomics/BGNN-trait-segmentation · Hugging Face
Container: Package bgnn-trait-segmentation · GitHub
Repository: https://github.com/hdr-bgnn/BGNN-trait-segmentation/tree/0.0.7|Maruf and Karpatne (2022)|
|Trait extraction: Morphology_analysis|This paper|hdr-bgnn/Morphology-analysis: Version 1.0.0
Container: Package morphology-analysis/morphology · GitHub
Repository: https://github.com/hdr-bgnn/Morphology-analysis/tree/v1.0.0|Tabarin, Bradley, Balk, and Lapp (2023b)|
|Other|
|Fish-AIR|https://fishair.org/|https://fishair.org/||
2.2.1 Image data and metadata
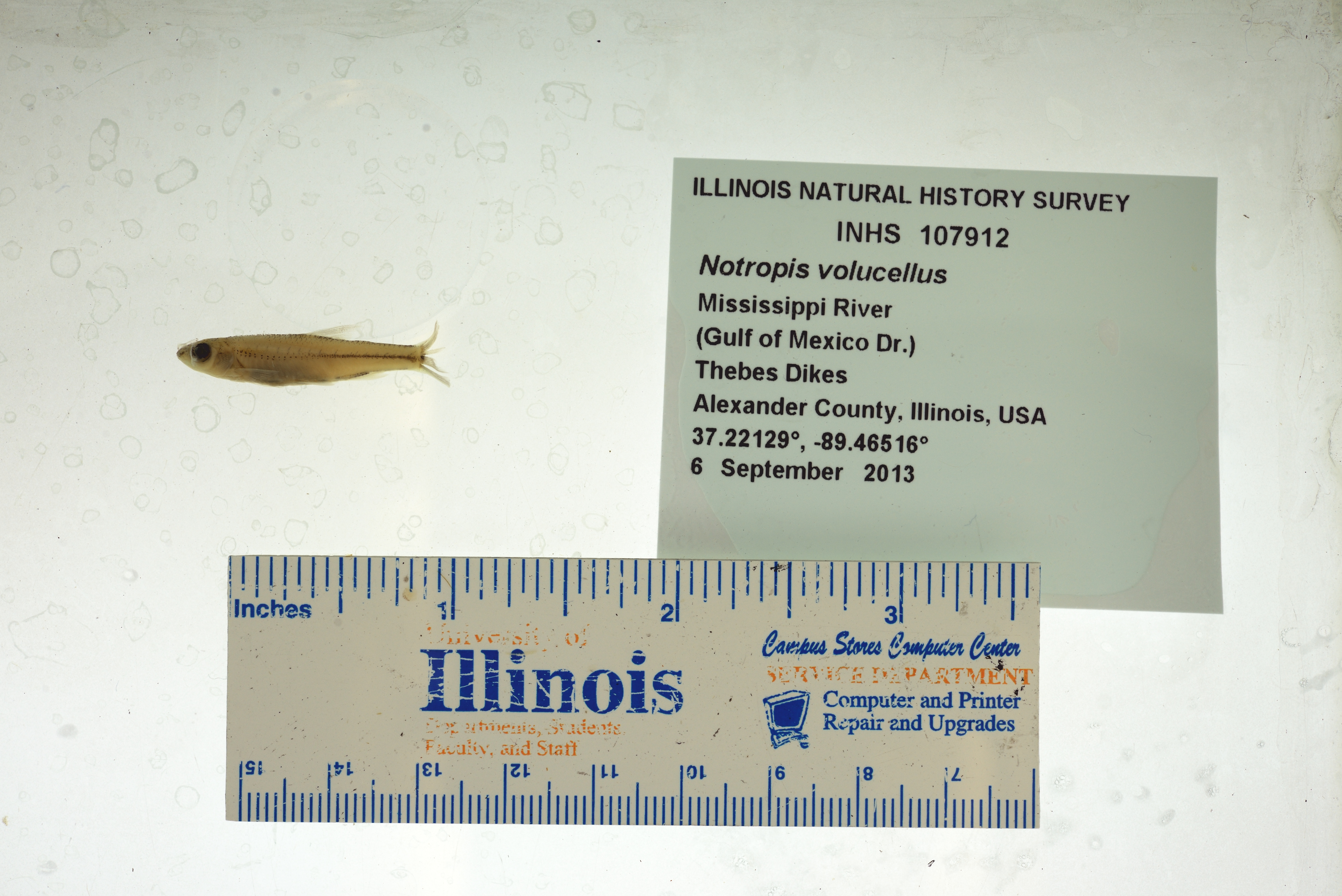
The case study uses image data generated by museums, specifically of freshwater museum fish specimens belonging to the minnow group (Family: Cyprinidae). The image data and metadata are hosted and downloaded from the Fish-AIR repository (https://fishair.org/), and were originally from the Illinois Natural History Survey (INHS; https://inhs.illinois.edu/) as part of the Great Lakes Invasives Network (GLIN; https://greatlakesinvasives.org/portal/). The associated extended image metadata (IMD) and image quality metadata (IQM) were extracted using the workflow described at Fish-AIR (FISH-AIR - WORKFLOW). The IMD includes information about image size and IQM includes qualitative information about the contents of the image (Leipzig et al., 2021). These metadata files that serve as the input for the automated workflow can be found in the folder ‘Files’ in the Minnow_Segmented_Traits repository (Table 1). The metadata files contain resolvable URLs to access the image data (Figure 2, metadata).
Maintaining the images and their metadata in a repository facilitates findability and accessibility of the image data and metadata by all members of a collaborative team (Brito et al., 2020; Goble et al., 2020; Miura & Nørrelykke, 2021). As a repository, Fish-AIR also facilitates the retention of provenance and attribution metadata of the image data (Table 1; Brito et al., 2020), which could easily be lost if sharing a local folder of images. To provide open and long-term access, and to ensure reproducibility of filtering steps (see below), we deposited the metadata files from Fish-AIR in a Zenodo archive (Table 1). For the image data, Fish-AIR as a repository provides open access to them under stable unique identifiers, even if it is currently not set up as a permanent archive.
2.3 Workflow manager
A workflow manager (WM; Figure 2, row 4) is a software tool for executing the steps in a computational workflow that is codified in the WM’s definition language. Ideally, the WM can: invoke the components, identify when a change has been made to re-complete a step, identify when a step has already been completed and not duplicate the work, and run the steps sequentially or in parallel. This automation afforded by the WM also helps prevent duplication of outputs and avoids missing critical steps (Brito et al., 2020; Goble et al., 2020; Sandve et al., 2013).
We use Snakemake as the WM, while acknowledging that there are many options (Wratten et al., 2021). Snakemake is well-suited for an image-based, collaborative application because it permits extensive documentation, is compatible with using HPC environments, is open source, requires relatively little setup, and is built on Python, a programming language already commonly used in image-based ML. Further, it is compatible with R programming, which is widely used in ecological, biodiversity, and evolutionary analyses, and it is therefore the language of choice for the domain scientist in our application. Additionally, Snakemake enables modularization of a workflow through user-defined rules or steps. Snakemake also allows for the specification of component versions, ensuring reproducibility and flexibility with testing changes to the codebase. Finally, Snakemake also generates log files, which are useful for debugging problems, reading errors, and for a domain scientist to work with a software engineer. Thus, the advantages are that the domain scientist can select which parts of the workflow to rerun and is empowered to troubleshoot (Roach et al., 2022).
Snakemake rules (Figure 2, row 4), which correspond to steps in the conceptual imageomics workflow, specify the commands that transform inputs into outputs by calling a component, such as executable programs, scripts, and containers (Köster & Rahmann, 2012). To more clearly link the generated output files to rules, we devised a naming convention for the outputs, ‘ARKID_ruleName.fileExtension’ (Sandve et al., 2013). We reduce redundancy and the potential for errors by using a configuration file that defines paths, file names, etc. that can be used by the workflow definition and custom scripts. Thus, if a path or file name changes or is added, the change needs to be made only in a single place, rather than repeatedly throughout (Roach et al., 2022). We leverage Snakemake’s capability to use entire workflows as components by creating a sub-workflow, BGNN_Core_Workflow (Table 1; Goble et al., 2020). This workflow consists of steps that are used by the entire collaborative team, not specific to a project, such as downloading image metadata and image data, generating and reformatting image processing metadata, creating a mask, cropping the image and applying the segmentation module.
2.3.1 Environment
Most workflows will require the creation of a computational environment suitable to run the various scripts and containers (Figure 2, create environment). We use a high-performance computing (HPC) environment to isolate the environment and allow multiple users to run the workflow. Our workflow requires Conda (Anaconda Software Distribution, 2020; for Python and Snakemake) and Singularity (now Apptainer; Kurtzer et al., 2017; Singularity Developers, 2021; to create and run Docker container images). The configuration file (config/config.yaml) sets the inputs and outputs as relative paths, as this allows paths to components or outputs to be changed only in the configuration file rather than multiple times across components, following best practices (Roach et al., 2022; Sandve et al., 2013). We also create YAML files to load environments, such as an R environment and for image data downloads, following best practices for version control and defining paths to components (Roach et al., 2022; Sandve et al., 2013). These files are in the folder ‘envs’ and called by the workflow definition.
We created a way to recreate the R environment used by domain scientists’ scripts, which were in the R programming language (version 4.2.3; R Core Team, 2018). We intentionally did not containerize the R dependencies and environment for ease of use for domain scientists who may not have expertise in containers. Instead, we supply a Conda environment YAML file (envs/r-minnows.yaml) so that Snakemake will automatically create an R environment before running the R scripts. The environment is initialized using init.R in the folder ‘Scripts’ by defining the paths to all files (paths.R) and initializing all the functions (functions.R). The computational environment automatically loads the R programming dependencies [dplyr 1.0.8 (Wickham, 2023a), ggplot2 3.3.5 (Wickham, 2016), ggpubr 0.4.0 (Kassambara, 2023), json 0.2.21 (Couture-Beil, 2022), moments 0.14.1 (Komsta & Novomestky, 2022), RColorBrewer 1.1.2 (Neuwirth, 2022), remotes 2.4.0 (Csárdi et al., 2022), reshape2 1.4.4 (Wickham, 2022), stringr 1.4.0 (Wickham, 2023b), tidyr 1.2.0 (Wickham et al., 2024), yaml 2.3.5 (Garbett et al., 2023)] into a created folder, ‘Library’.
2.4 Components
Components are the scripts and tools, such as containers and ML models, that the WM invokes based on the rule definitions (Figure 1, row 2; Figure 2, row 3). We store, build, and develop the components using GitHub for version control and for making the components findable and accessible to the full team (and the public). The components used are either an entire repository that is later containerized or scripts within the project repository. We created specialized components for this case study, though general to our collaborative team and thus can be used as modular, interoperable components in any future workflow.
Containerizing the components enables interoperability and portability for use in a workflow (Brack et al., 2022; Gruening et al., 2018; Nüst et al., 2020; Roach et al., 2022). Although the trained models can be included directly in the codebase, this makes individual components difficult to identify and access, inhibiting reuse. Therefore, we consider models (more specifically, the trained ML model weights) as their own digital objects, and deposit them in Hugging Face (https://huggingface.co/) where they receive their own identifier and resolvable URI, and from where they can be downloaded by the component (Gruening et al., 2018; Kadri et al., 2022). A domain scientist can incorporate as many of these components as necessary for their project. We chose to containerize components using Docker (Merkel, 2014) as these containers are compatible with Singularity, and therefore Snakemake and most HPC environments. Below we discuss the specific components in our case study and their implementation into the conceptual imageomics workflow.
2.4.1 Download metadata
To be completely automated, the first step is to read in the metadata (Figures 1 and 2, step 1)—that is, the metadata is not stored in the Minnow_Segmented_Traits repository (rules download_fish_air_data or download_zenodo_data). The IMD provides a unique ID [called ARKID by Fish-AIR, but as a current limitation of Fish-AIR they lack the Name Assigning Authority Number (NAAN) prefix and are thus not resolvable as ARK IDs] and path to download for each image datum. This step downloads IQM (imageQualityMetadata.csv), which contains information about each image, such as if the specimen is curved or obstructed in the image. Since we restrict image data to contain species that overlap with those in Burress et al. (2017), the WM invokes the rule download_burress to download Burress et al. (2017) supplementary data for later image filtering (see Section 10).
2.4.2 Filter image data
The specific filtering of the image data and metadata are unique to our case study; however, the implementation of this step is generalizable (Figures 1 and 2, step 2). The filtering scripts are executed by rule select_minnow_images. We first manipulated the metadata files for ease of use using R scripts. We created a custom script, Data_Manipulation.R in Scripts to combine the IQM and IMD files, and to modify the Burress et al. (2017) supplementary file for future downstream analyses. To identify the image data to download, we again used a custom script, ‘Minnow_Selection_Image_Quality_Metadata.R’ in ‘Scripts’. High-quality minnow image data were selected based on the parameters and values recommended by Leipzig et al. (2021) (Table 2). Finally, we selected only those species that were also in Burress et al. (2017). This resulted in a final dataset of 13 species and 273 image data records (Table S1).
TABLE 2. Filtering criteria for Minnow image download using Fish-AIR vocabulary (FISH-AIR - VOCABULARY).
| Statement | IQM | Options | Filter | Example of a removed image: |
|---|---|---|---|---|
| If the specimen is not curved | specimenCurved | straight, curved | Remove all images of specimens that are not straight | https://fishair.org/hdr-share/ftp/ark/89609/GLIN/UWZM/1p85b80p.jpg |
| If all the parts of the fish are visible | allPartsVisible | True, False | Remove all images of specimens that have parts not visible | https://fishair.org/hdr-share/ftp/ark/89609/GLIN/INHS/1105vw55.jpg |
| If no objects overlap with the fish | partsOverlapping | True, False | Remove all images of specimens where fins or other parts overlap | https://fishair.org/hdr-share/ftp/ark/89609/GLIN/INHS/2s96p382.jpg |
| If the fins are folded | partsFolded | True, False | Remove all images of specimens where fins or other parts are folded | https://fishair.org/hdr-share/ftp/ark/89609/GLIN/UWZM/9f80858x.jpg |
| If the background is uniform | uniformBackground | True, False | Remove images where background is not uniform and so may obstruct machine learning tools | https://fishair.org/hdr-share/ftp/ark/89609/GLIN/INHS/sh31rb7q.jpg |
| Fish facing left | specimenView | left, lefttop, leftbottom, right | Remove images of specimens which are not facing left | https://fishair.org/hdr-share/ftp/ark/89609/GLIN/UWZM/1p72wx0g.jpg |
| Fish possessing all parts | partsMissing | True, False | Remove images of specimens missing fins or other parts | https://fishair.org/hdr-share/ftp/ark/89609/GLIN/INHS/xv05jh70.jpg |
| Brightness | brightness | normal, dark, bright | Remove images either too bright or too dark | https://fishair.org/hdr-share/ftp/ark/89609/GLIN/INHS/1942xz57.jpg |
| Focus | onFocus | True, False | Remove images that are not in focus | https://fishair.org/hdr-share/ftp/ark/89609/GLIN/INHS/q794d840.jpg |
| Color | colorIssue | none, contrast, slightly dark, very dark, dark, dark spots, small black dots on posterior of fish, blotchy discoloration, specimen is dark, light spots, slightly dim and dirty picture, specimen has discoloration, discoloration on scales, anterior portion, dim | Remove images with color issues | https://fishair.org/hdr-share/ftp/ark/89609/GLIN/INHS/6p98d534.jpg |



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
https://fishair.org/hdr-share/ftp/ark/89609/GLIN/INHS/xx32hs1k.jpg|
|Ruler included|containsScaleBar|True, False|Remove images which do not have a scale bar|All cases were true; we checked that the scale bar was perceived by the object detection.|
- Note: The option kept is bolded. Fish image unique IDs are linked to their image hosted by Fish-AIR.
2.4.3 Download image data
Image data are downloaded from the Fish-AIR repository based on a unique URL from a file in the Zenodo archive (Figures 1 and 2, step 3; Table 1). This is encoded in the rule download_image from the BGNN_Core_Workflow. These image data are stored locally in a new folder ‘Images’ for further processing. Storing data locally rather than on the shared GitHub repository helps keep the repository size down. All image data are saved as ARKID.jpg, where ARKID is an unique identifier assigned to the fish image data by Fish-AIR.We added a limiting step in the downloading component so that the domain scientist can specify the number of image data to be downloaded in the ‘config.yaml’ file. This helps with testing, as the domain scientist can select 10 image data, as an example, speeding up processing time (Roach et al., 2022). The input is an integer or to download all the image data (‘’) for the final runs.
2.4.4 Metadata generation
The first ML component (Figures 1 and 2, step 4a) performs object detection and metadata generation as defined by the rule generate_metadata from the BGNN_Core_Workflow (Tabarin, Bradley, Balk, & Lapp, 2023a; Table 1). This rule calls the container drexel_metadata from the drexel_metadata repository (Karnani et al., 2023; Table 1), which generates a metadata file for each image datum (named ARKID.json). The codebase has two outputs and is described in Leipzig et al. (2021), Pepper et al. (2021), and expanded upon in Karnani et al. (2022). The domain scientists and software engineer worked with the ML researchers to containerize their codebase for reuse in this workflow.
This component uses detectron2 (He et al., 2018; Wu et al., 2019) to detect five instance classes within images: fish, fish eye, ruler, number ‘2’, number ‘3’. detectron2 outputs bounding boxes, binary masks, and a series of attributes for each instance of each class. The generate_metadata software post-processes information using the instance classes, like determining the image scale (pixels/cm). If a fish is identified, generate_metadata creates a new, refined binary mask over the specimen. These masks are created by using the initial bounding box from detectron2 and performing pixel-based contrast analysis to determine the contours of the fish. The bounding box is recalculated based on this refined mask. These image data are stored in the folder ‘Mask’ and are named ARKID_mask.png. The final bounding boxes created are used further down the pipeline to crop the fish specimen from the rest of the image so that the tag and ruler are not fed into the segmentation model.
2.4.5 Reformat metadata
We created a component called drexel_metadata_formatter (Tabarin, Bradley, & Lapp, 2023a; Table 1) that reformats the output from the generate_metadata component to only include what is needed for downstream analyses for this case study. This rule, transform_metadata, is from the BGNN_Core_Workflow that the WM calls. We retain base_name, version, fish number, bbox, pixel_analysis, rescale, eye_bbox, eye_cener, angle_degree, eye_direction, foreground_mean, background_mean, ruler, and ruler_bbox, ruler_scale. These intermediate data are stored locally in the folder ‘Metadata’. This is an example of a domain scientist customizing the component. In this example, metadata can be reformatted or scripts in R or Python to reformat in a domain scientist’s repository that are not containers.
2.4.6 Crop image
We cropped the image data to only include the fish; extraneous items such as the specimen tag and scale bar were removed. To implement this step in a reusable way, we added code for cropping image data in its own repository, Crop_Image (Tabarin, Bradley, & Lapp, 2023b; Table 1), which made it easy to containerize. The corresponding rule, crop_image, is invoked by the WM from the BGNN_Core_Workflow. Using the output from the generate_metadata rule, we increased the bounding box by 5% (2.5% per side) to crop the image. These image data are stored in the folder ‘Cropped’ as ARKID_cropped.jpg. This is an example of the domain scientist interacting with the ML researcher’s products.
2.4.7 Segmentation
The second ML component is a segmentation model (Figures 1 and 2, step 4b). To facilitate integration and improve reusability, we containerized the segmentation model. The Docker container image includes the code (model architecture, preprocessing, and post-processing code) and downloads the model weights from the BGNN-trait-segmentation (Maruf & Karpatne, 2022; Table 1), to build a full-fledged independent tool for the segmentation (Table S3). The segmentation model is from the Segmentation Models library (Iakubovskii, 2019), which is based on PyTorch (Paszke et al., 2017). The model was previously trained using ImageNet (Deng et al., 2009), then fine-tuned to our dataset (see Data S1). The segmentation model classifies pixels on the image of the specimen as a trait (Figure S1). The results of the segmentation model are in the folder ‘Segmentation’ and the image data are stored as ARKID_segmented.jpg. These results are then used for downstream analyses.
We modified the codebase, BGNN-trait-segmentation (Maruf & Karpatne, 2022; Table 1), which is invoked from the BGNN_Core_Workflow, to work with our images. Since segmentation models are designed to process a specific image size, we needed to resize the cropped image data to minimize spatial distortion. The resizing of images was based on the size distribution of the images in the dataset and resizing to the mean of each dimension’s distribution. We did not add any padding to the images. After segmentation, the image is again resized to the size of the cropped image so that the scale remains the same as the original image data from which the ruler has been extracted. We worked with the ML researcher to containerize their codebase for reuse in this workflow.
2.4.8 Morphological analysis
We created two components to analyse segmentation outputs (Figures 1 and 2, step 5). One component extracts the classification of the traits (i.e. segmentations) from the segmentation model. We created a codebase, Morphology-analysis (Tabarin, Bradley, Balk, & Lapp, 2023b; Table 1), for scripts to record the number of groupings of continuous areas of pixels classified with the same label, which we call blobs, corresponding to a trait (Figure S2), the percentage by area (number of pixels) of the biggest blob. The container is invoked in the rule create_morphological_analysis in the workflow. The outputs are saved as a json file, ARKID_presence.json in ‘Presence’.
The other component is a R script for statistical analysis. In general, the nature of the statistical analysis on the ML-generated outputs will be specific to a domain scientist’s research question(s). For our case study, we created a script, Presence_Absence_Analyses.R in ‘Scripts’, for deriving presence or absence metrics of the traits segmented by the Segementation step. The rule presence_absence_analysis in the workflow invokes this script and creates figures and tables that are put into the folder ‘Results’. In general, domain scientists require creative control over this component, and thus writing a script to be stored in the project repository is appropriate.
We combine the ARKID_presence.json files (Table S2). We then performed analyses on how well the model performed at finding the traits and created visualizations that are stored in ‘Results’. Although the segmentation performance was high [mean IoU of 0.9 (scores range from 0 to 1)], some artefacts were present such as the erroneous presence of traits or fragmented trait segmentation. We assessed the degree of uncertainty in identifying a trait by quantifying how many traits had the biggest blob as less than 85% of total trait blobs and the spread of the area of the biggest blob for each trait (Figure S3).
3 RESULTS
The conceptual imageomics workflow is built on the principle of modularity; the application-specific imageomics workflow is successfully implemented and executed following FAIR principles (Table 3; Barker et al., 2022; Goble et al., 2020; Wilkinson et al., 2016). For all data, metadata, software, and ML models used, we created rich metadata files, retained provenance and attribution, store and retrieve from findable, accessible repositories, registries or archives. To achieve findability and accessibility we referenced repositories for metadata and components in the workflow definition enabling the automatic download of image data and execution of the workflow rules. To create modular and portable components we used containers and specified component versions for reproducibility. To automate the workflow, we utilized a WM. We use configuration files to define relative paths for both the Snakemake workflow definition, the scripts invoked by the WM, and for setting up the environment, thereby reducing redundancy. Using the WM with a configuration file facilitated interoperability of all components and scripts.
TABLE 3. Table showing the ways our workflow components adhere to the FAIR principles.
Principle Levels Data and metadata Software ML models
Wilkinson et al. (2016) Our application Barker et al. (2022) Our application Our application
Findable 1 Data and metadata are assigned a globally unique identifier All metadata and data are available with the archives, which have a globally unique identifier (Table 1) Assigned a globally unique identifier All software have a globally unique identifier for their archive (Table 1) ML models are given a unique identifier and are versioned (Table 1)
1.1 Components of software representing levels of granularity are assigned distinct identifiers Every release is given a new DOI by the archive Revision included in DOI citation
1.2 Different versions of the software are assigned distinct identifiers Each version is given a unique identifier using Semantic Versioning Identifiable by commit hash from version control
2 Data are described by rich metadata All data items have associated metadata with the archive and in their repository Described by rich metadata All software have associated metadata in the archive and in their repository All models have associated metadata in the repository in the form of rich model cards
3 Metadata clearly and explicitly include the identifier of data or software they describe Metadata includes provenance for training and base models
4 Data and metadata are registered in a searchable resource All image metadata are hosted by Fish-AIR, which is searchable, and they are in an archive; all image data are hosted by Fish-AIR, which is searchable, and retrievable from their metadata files Metadata are FAIR, searchable, and indexable Software container images are in the GitHub container registry Models are in Hugging Face repository, which is searchable by various model attributes
Accessible 1 Data and metadata are retrievable by identifier using a standardized communications protocol We bring in metadata and data from the archive using its unique identifier and import using WM and config file Retrievable by identifier using a standardized communications protocol Code from GitHub and container images are recruited using unique identifiers and container version by the workflow definition Models and their weights are retrievable by a unique identifier by the workflow definition
1.1 Protocol is open, free, universally implementable Fish-AIR (repository) and Zenodo (archive) use HTTP(S) GET Protocol is HTTPS or Git version control
1.2 Protocol allows for authentication and authorisation, when necessary
2 Metadata are accessible, even when data or software are no longer available We have created a download to retrieve data and software
Interoperable 1 Data and metadata use a formal, accessible, shared language for knowledge representation Metadata in a csv file that is readable; image data are retrieved from a URL Reads, writes, and exchanges data in a way that meets domain-relevant community standards We read in and use the software through a WM that is available to the community and used in biological research Models are in PyTorch format
2 Data and metadata use vocabularies that follow FAIR principles Vocabularies for metadata determined by the community (for the fish image metadata) or are from Darwin Core (DwC; Wieczorek et al., 2012), Dublin Core (DC; Caverlee et al., 2018), and Audiovisual Core (AC; via the Biodiversity Information Standards) (TDWG; https://www.tdwg.org/)
3 Data and metadata includes qualified references to other data and metadata Metadata references the image data by URL; some metadata vocabularies from DwC Includes qualified references to other objects Software dependencies enumerated in requirement definitions (i.e. for python, R, external repositories, model weights, and all container images).
Reusable 1 Data and metadata are richly described with a plurality of accurate and relevant attributes Fish-AIR has image metadata and vocabulary documentation Richly described with a plurality of accurate and relevant attributes Software metadata includes authors/attribution, licensing, release version and data, mention other dependencies where necessary, and rich, informal descriptions (e.g. README files) Rich metadata descriptions in the form of model cards, includes authors/attributions, licensing, and versioning in the form of commit hashes
1.1 Data and metadata are released with a clear and accessible data usage licence All components, repositories, and archives have a licence Released with a clear and accessible data usage licence All software, repositories, and archives have a licence All models have a licence
1.2 Data and metadata are associated with detailed provenance All image metadata include information of the hosting and data-governing institution Associated with detailed provenance All repositories, container images, and archived snapshots include information about authors and versioning; all GitHub repositories have CITATION.cff files Metadata describes the software and data used to train the models to produce the weights
1.3 Data and metadata meets domain-relevant standards Image metadata format and vocabulary use domain-relevant standards (i.e. CSV, XML, DwC); image data use standard formats Meets domain-relevant standards Container images are dockerized and reusable by Singularity in HPC environments; YAML files define code dependencies Model cards are a Hugging Face standard, and PyTorch is a ML standard
- Note: Note that to the best of our knowledge there is not yet a reference publication for FAIR standards for ML Models, however guidance is being developed by the FAIR4ML Interest Group within the Research Data Alliance (FAIR for Machine Learning (FAIR4ML) IG Members).
Our inputs and outputs are findable and accessible. Our metadata files, data items, software components, and ML models are assigned unique identifiers and are stored in a searchable resource. Further, the metadata files and image data are retrievable using their identifiers; this accessibility enables reproducibility by the public. We take advantage of the built-in metadata structure and attributions when depositing data (Grossman et al., 2016). Beyond the metadata, the workflow uses technology that is FAIR, such as Snakemake, GitHub, and the containerized components, all which are accessible and free to use.
To achieve interoperable and reusable components that are portable across HPC or computing platforms, we create Docker container images for each component’s codebase. We fully automated the potentially tedious process of updating container images for a code repository upon updates, using GitHub Actions that both build a Docker image (i.e. container) and then push it to GitHub’s Container Registry (https://ghcr.io) automatically with every code release. Being part of a registry of containers ensures that these components are findable and accessible, with associated metadata and data items, so that the provenance of the source of the container is apparent. We also archive the code repositories for perpetuity to facilitate future reproducibility (see ‘Data Availability Statement’). We follow best practices of containerization, such as limiting one tool (or component) per container, including licensing, ensuring container accessibility and keeping the data separate (Gruening et al., 2018).
Versioning code and components was critical for reproducibility and project development. For this we followed Semantic Versioning (https://semver.org) where a versioning scheme was not already present in a component’s repository, by applying corresponding Git version control tags to respective commits. To allow for flexible control of which version of a component is recruited in the workflow, we exploited the fact that the same container image in a registry can have multiple tags representing different version granularities (Nüst et al., 2020), meaning that the domain scientist can select a specific version or the latest version.
4 DISCUSSION
Harnessing data in the form of images is dramatically increasing across the biological sciences. New tools, models, and algorithms for analysis are being rapidly produced and recombined to address an increasing variety of research questions. We posit that for this emergent field to develop its full potential in facilitating novel research endeavours involving interdisciplinary teams, it is important to establish FAIR data and reproducible workflow practices (Barker et al., 2022; Brack et al., 2022; Goble et al., 2020). All team members need to be able to find and access the data, models, and any components, which usually means that the corresponding workflow ingredients need identifiers and be publicly available. The research-grade tools developed by team members needed to be made interoperable, reusable, and portable for use across computational environments, and in other projects, which we accomplished by containerizing tools and placing the container images into a public registry. Further, unlike data simply comprising facts of nature, image data requires special attention to best practices with respect to attribution, provenance chain and licence.
We tackle how to combine tools and technologies emerging from active research for using ML to process and extract knowledge from image data such that the resulting workflow is end-to-end automated, reproducible and (re)usable by domain scientists. We start by developing a conceptual imageomics workflow and then create an application-specific implementation emphasizing FAIR principles, modularity, portability, and flexibility. The application of the conceptual imageomics workflow is intended to serve as a guideline for team implementation of a workflow incorporating image data and ML tools using FAIR data principles. By focusing on these principles and techniques, we avoid common pitfalls when trying to use another researcher’s tool, such as code redundancy and duplication, failure-prone dependency management, non-replicable computing environments, or non-portable file paths. Developing such a workflow template facilitates teamwork as its implementation requires deep collaboration among the ML researchers, domain scientists and software engineers. In this sense, some of the benefits from the template and approaches we describe will most directly accrue to an interdisciplinary team that includes software engineering expertise, and less so to an individual domain scientist or ML researcher.
4.1 Team science
In our team, we recognized that it is important to provide and retain author attribution for all scripts and inputs so that all member contributions are valued and appreciated. While such documentation may seem mundane, we found attribution is best addressed early in the project to bring clarity about who is working on the code and to incentivize collaboration. Giving appropriate credit not only made team members feel valued, it also served as a point of contact should the domain scientist need help. Attribution was provided in multiple places: with data items or trained model weights in a repository, with any scripts in a repository, and with the container image in a registry. Acknowledgements, licensing and attributions were provided in the readme and as a script header. Adding licensing makes the rights and terms of reuse clear for each component (Goble et al., 2020). Accessible metadata facilitates reproducible and reusable science (Brito et al., 2020).
It is a common observation that code artefacts from active computational research often fall under what is referred to as research-grade, in contrast to product-grade robust and reusable tools (Grüning et al., 2019; Trisovic et al., 2022). We, too, encountered interoperability issues for command-line invocation of components, unstable expectations for inputs and outputs, and other problems when ML tools under active research and development needed to be included in the larger workflow. We argue that this will be encountered commonly in imageomics, given that ML components will remain under highly active and dynamic research. We used Semantic Versioning tags for the containerized components enabled the domain scientist to precisely control which version of corresponding ML codes, models, and associated software dependencies is used in their workflow, enabling reproducibility (Brack et al., 2022; Goble et al., 2020; Niehues et al., 2024; Nüst et al., 2020; Roach et al., 2022). Snakemake allows specifying the desired tag for a container and also recognizes if the container for a step (‘rule’) is of a different version than the one with which the step has run previously, and, if so, re-runs the rule.
4.2 Workflow
The research and development of WMs is a very active endeavour, and consequently there are many choices. We found that our core technical requirements, such as modularity, automation, and HPC interoperability, are met by many candidate WMs, but that usability by team members can differ substantially. In our case, we chose Snakemake as the WM that provided usability by a domain scientist with basic familiarity with image data science using Python tools and has capabilities for using containers, which is how the modules are implemented. For the software engineers and ML researchers on the team, Snakemake’s rule system is conceptually similar to rules in Makefiles. This choice has also been reached elsewhere in a related context (Schmied et al., 2016).
The replicability of a workflow by different users in a team, or on different computing environments, necessarily hinges on the files required for any given step to be findable by the components. Initially, our files were maintained on a local filesystem, and hence the paths to access these files were at first hardcoded, rendering them non-portable. We addressed this by downloading input files from a repository on-demand, and by removing all hard coded paths, and by defining file paths and identifiers in a configuration file for the workflow. As a side benefit, this configuration file also allows us to set options specific to a computing environment, such as whether a GPU is available to the ML components or not.
Similar to genomic data analysis, image data analysis can consist of conditional steps, branching, and loops, but in the absence of a formal workflow definition for end-to-end automation the corresponding computations often run without coordination, parallelization, and orchestration. Automation of workflows thus promotes modularity and efficiency. Assembling all computational components in a study into an automated workflow is also key to reproducibility. Workflows do not need to begin fully modularized or even automated; a monolithic and manually-operated approach may even be more productive in the early stages of an analysis project. For example, our workflow began as manual because it required less upfront technical effort and fewer unfamiliar technologies to learn during the exploratory stages, when the team was still determining the necessary inputs, the data wrangling steps; and the desired outputs. As more team members needed to run all or part of the analysis, automating components of the workflow brought significant time savings throughout the continuation of our project and enabled reuse between the domain scientist, ML researchers and software engineers. Assembling all computational components in a study into an automated workflow is also key to its full computational reproducibility.
4.3 Modularity
Machine learning research can result in multiple tools being maintained in a single large and therefore monolithic repository that performs many tasks. Modularizing these tools as workflow components made our workflow easier to understand, automate and therefore use. Further, modularization via components allowed for faster prototyping of a project because elements can be ‘plug and chug’; a simple change in the workflow could be tested immediately. The downside of modularization is keeping track of external dependencies and versions for each component. Still, we found that modular components also promoted reuse by our domain scientist and improved computational efficiency.
We adopted earlier best practices for allowing domain scientists to use and combine research-grade software tools created by dynamic collaborative research endeavours, such as the one we report here, into an automated and reproducible workflow (Leipzig et al., 2021; Shade & Teal, 2015). Specifically, we use Docker containers to allow packaging, distributing, and running research-grade code in a reproducible environment, isolated from the other tools in a workflow. Containerization that follows FAIR principles also prevents codebase duplication, the loss of associated metadata, and makes components portable (Nüst et al., 2020). Although Docker is typically unavailable in a shared HPC environment due to the elevated privileges it requires, Singularity (now Apptainer), which runs on a Linux operating system and is usually supported on HPC clusters, can bootstrap its containers from Docker container images. Further, automating the process of container image creation and versioning via GitHub Actions made reproducing the software environment easy for the domain scientist. Our collaborative team had the benefit that the ML researchers who originally built the tools were a part of the team and aided in containerization of these components. Not all teams may have this benefit, especially when using third-party tools. It is important, nonetheless, to be transparent on which components are reusable and which are not for future use.
5 OUTLOOK
The future of imageomics is an exciting one: more tools, more methods, more models will undoubtedly be developed. With these developments, the need to bring them together to build fully automated and reproducible workflows will become ever more prevalent. The technologies and practices used to realize the conceptual imageomics workflow into an application-specific, automatic and reproducible workflow will serve as a guide that reduces tool wrangling and frees time to explore scientific questions.
AUTHOR CONTRIBUTIONS
Meghan A. Balk, Hilmar Lapp, Paula Mabee, Christopher R. Florian, Thibault Tabarin conceived the ideas; Meghan A. Balk, Thibault Tabarin, Christopher R. Florian, Hilmar Lapp and John Bradley designed the methodology; Dom Jebbia developed the metadata and standards management framework, and Yasin Bakis, Henry L. Bart, Jr., Bahadir Altintas, Xiaojun Wang made image data and metadata available through Fish-AIR; Joel Pepper, Jane Greenberg, Kevin Karnani, David Breen designed the objected detection model; M. Maruf and Anuj Karpatne designed the segmentation model; Meghan A. Balk, Thibault Tabarin analysed data; John Bradley, Thibault Tabarin, Meghan A. Balk, Hilmar Lapp and Christopher R. Florian designed the workflows; Meghan A. Balk, Hilmar Lapp and Paula Mabee led the writing. All authors contributed critically to the drafts and gave final approval for publication.
ACKNOWLEDGEMENTS
We thank the Illinois Natural History Survey (INHS) for providing the image data used in this study. We thank W. M. Dahdul and K. Diamond for their time and expertise in annotating fish image data. We also thank E. Campolongo for metadata help.
FUNDING INFORMATION
We thank the NSF-funded Imageomics Institute (NSF 2118240: HL, JB, PM, CF, TT, MAB) led by T. Berger-Wolf for their support and other NSF funds including NSF 2022042: PM, CF, TT, MAB; NSF 1940233: JG, JP, KK, DB; NSF1940322: HB, YB, BA, XW, DJ; NSF 1940247: AK, MM.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflict of interest.